

Motif prediction to distinguish LPS-stimulated pro-inflammatory vs. antibacterial macrophage genes
- Research
- Open Access
Motif prediction to distinguish LPS-stimulated pro-inflammatory vs. antibacterial macrophage genes
- Rahul K Kollipara1_37 and
- Narayanan B Perumal1_37Email author
- Received: 7 September 2010
- Accepted: 21 September 2010
- Published: 21 September 2010
Abstract
Background
Innate immunity is the first line of defence offered by host cells to infections. Macrophage cells involved in innate immunity are stimulated by lipopolysaccharide (LPS), found on bacterial cell surface, to express a complex array of gene products. Persistent LPS stimulation makes a macrophage tolerant to LPS with down regulation of inflammatory genes ("pro-inflammatory") while continually expressing genes to fight the bacterial infection ("antibacterial"). Interactions of transcription factors (TF) at their cognate TF binding sites (TFBS) on the expressed genes are important in transcriptional regulatory networks that control these pro-inflammatory and antibacterial expression paradigms involved in LPS stimulation.
Results
We used differential expression patterns in a public domain microarray data set from LPS-stimulated macrophages to identify 228 pro-inflammatory and 18 antibacterial genes. Employing three different motif search tools, we predicted respectively four and one statistically significant TF-TFBS interactions from the pro-inflammatory and antibacterial gene sets. The biological literature was utilized to identify target genes for the four pro-inflammatory profile TFs predicted from the three tools, and 18 of these target genes were observed to follow the pro-inflammatory expression pattern in the original microarray data.
Conclusions
Our analysis distinguished pro-inflammatory vs. antibacterial transcriptomic signatures that classified their respective gene expression patterns and the corresponding TF-TFBS interactions in LPS-stimulated macrophages. By doing so, this study has attempted to characterize the temporal differences in gene expression associated with LPS tolerance, a major immune phenomenon implicated in various pathological disorders.
Keywords
- Tolerant State
- Motif Prediction
- Identify Target Gene
- Transcriptomic Signature
- TFBS Prediction
Background
Innate immunity, one of the two arms of the immune system, provides the first line of defence against pathogens in mammals and nearly all other living things. Animals in the lower evolutionary scale, such as insects, fight off infections solely employing innate immune mechanisms. The innate immune system quickly alerts the host of the presence of microbial pathogens and this response is mediated through the expression of a limited number of receptors called pattern recognition receptors (PRRs) to identify pathogen associated molecular patterns (PAMPs) expressed by invading pathogens. Infections trigger PRRs, such as toll-like receptor (TLR) genes [1], to recognize PAMPs of invading pathogens and prompt an intracellular signalling cascade which ends in induction of pro-inflammatory cytokines, chemokines, type I interferons, and antimicrobial effectors that are essential for providing continuous protection from infection. In highly evolved organisms (starting from jawed vertebrates) innate immunity is required for priming adaptive immunity, the second arm of the immune system with long-term and specific immune response [2]. Macrophages are immune cells highly involved in conducting innate immune responsibilities and also play a role as an antigen-presenting cell (APC) to T lymphocytes (in adaptive immunity); they express various TLR genes to mediate the pro-inflammatory and antimicrobial responses.
LPS, a major component of gram-negative bacterial cell surface, is a potent stimulator of macrophages. LPS acts via the TLR4 receptor to trigger downstream signalling and expression of pro-inflammatory and antibacterial genes [1]. This induction needs to be under tight control since dysregulated inflammation can cause a number of pathological disorders such as septic shock, autoimmunity, atherosclerosis and cancer [3]. Various mechanisms of negative regulation of TLR-induced gene expression have been proposed to dampen uncontrollable inflammation [4] and these collectively lead to the phenomenon of "LPS tolerance" [5] wherein there is decreased expression of pro-inflammatory genes when there is prolonged LPS administration. Foster et al [6] have characterized the gene expression profiles of macrophages differentially treated with LPS to classify the genes into various phenotypic states including a tolerant state obtained by an initial LPS treatment. Their analysis of the genes expressed in the tolerant phenotype categorized the genes as belonging to "tolerizable" or "non-tolerizable" sets depending on no induction or further induction respectively during a second LPS treatment compared to the first one. Although LPS tolerance could prevent pathological inflammatory conditions in chronic bacterial infections, there is a strong need for a persistent antibacterial response to keep the infections under control. The set of genes that exhibit the tolerizable phenotype can be considered "pro-inflammatory" while those belonging to the non-tolerizable phenotype as "anti-bacterial".
Transcriptional regulation is a crucial biological mechanism controlling gene regulation in the tolerant phenotype vs. the basal state that corresponds to no LPS stimulation. A number of studies have looked at the transcriptional programs in LPS-mediated macrophage stimulation including the roles of TFs in prolonged LPS treatment [4, 7, 8, 9]. Roach et al [7] carried out a holistic approach to identify 92 TFs in human macrophages stimulated with LPS; however, this study did not distinguish between the tolerant and basal states since there was no re-stimulation with LPS in this study. A recent study characterized the Cebpd TF as a potential regulator of a switch between the basal and tolerant state [9]. A microarray analysis performed by Mages et al [10] similar to the Foster et al [6] experiment employed in our study, observed diminished gene expression of a vast majority of LPS-induced genes upon a second LPS treatment (tolerizable). However, these authors did not characterize any of the TF-target interactions responsible for the various phenotypes distinguished in their analysis. In order to achieve a global perspective on the transcriptional regulatory mechanisms inherent in LPS tolerance, we examined the Foster et al microarray data [6] for the control of pro-inflammatory vs. antibacterial gene expression. Using bioinformatics approaches, we characterized TF-TFBS interactions that differentiate LPS-stimulated pro-inflammatory vs. antibacterial gene expression depending on the well-accepted premise of coordinate expression corresponding to appropriate TF-TFBS interactions. Further, we show that some of these TF-TFBS interactions predicted from our analysis have been biologically validated as transcriptional targets in the literature with evidence of roles in LPS tolerance.
Results
Classification of antibacterial and pro-inflammatory macrophage genes
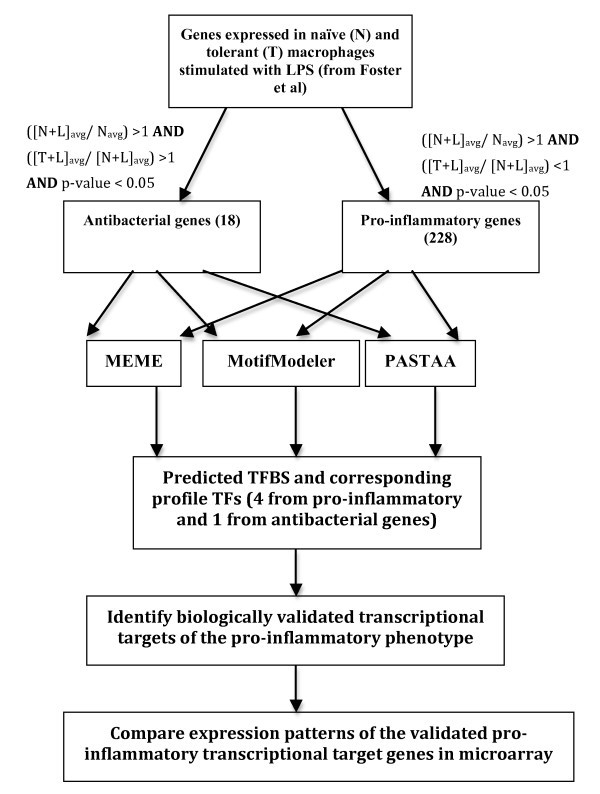
Workflow of experimental analysis to distinguish transcriptomic signatures of pro-inflammatoryvs. antibacterial genes. Average gene expression values in untreated macrophages, (N)avg, 4 hrs LPS-treated macrophages, (N+L)avg, and 24 hrs LPS-treated followed by 4 hrs re-treated macrophages, (T+L)avg were calculated. Two sets of genes were classified by the conditions mentioned in Methods.
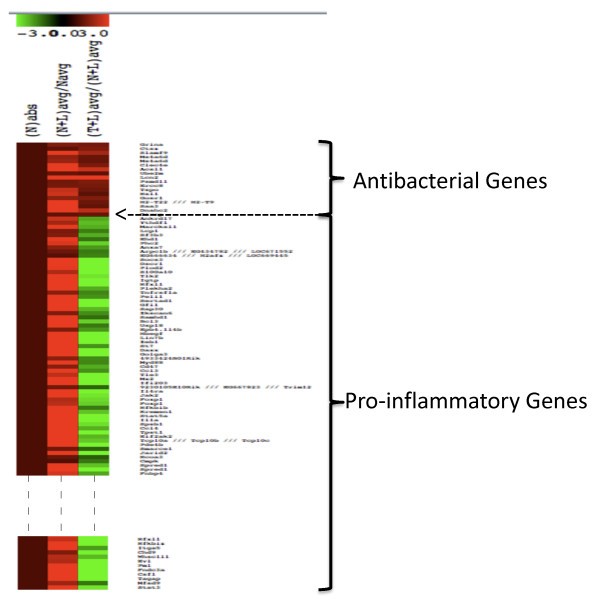
Heat map showing the differential expression patterns of LPS-stimulated genes. Navg, (N+L) avg and (T+L) avg are as described in Figure 1. Nabs was introduced to distinguish the patterns clearly and it is equal to one. The rationale was that the fold change is >1 for both classes of genes in the N+L phenotype, and <1 for pro-inflammatory and >1 for the antibacterial class genes in the T+L phenotype. Only a subset of the pro-inflammatory genes is shown.
Gene ontology (GO) analysis of the two gene sets
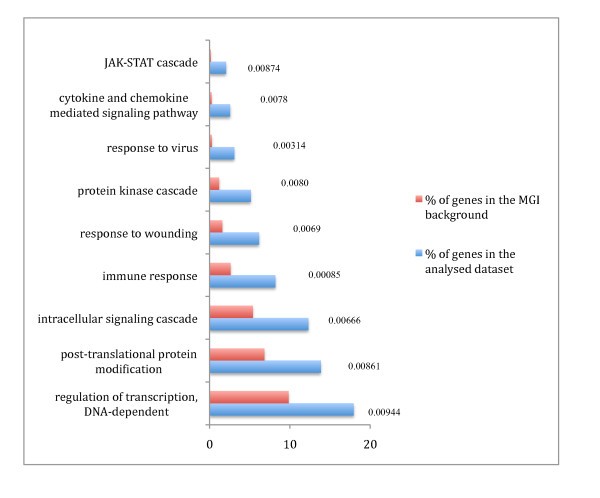
GO analysis of the pro-inflammatory genes. GO analysis of the pro-inflammatory genes based on annotated biological process GO terms was done using GOstat [37]. Enrichment of the pro-inflammatory gene set compared to a Mouse Genome Informatics (MGI) murine genome background d for each GO term is shown with significant p-values (< 0.01).
Characterization of TFBS in the two data sets
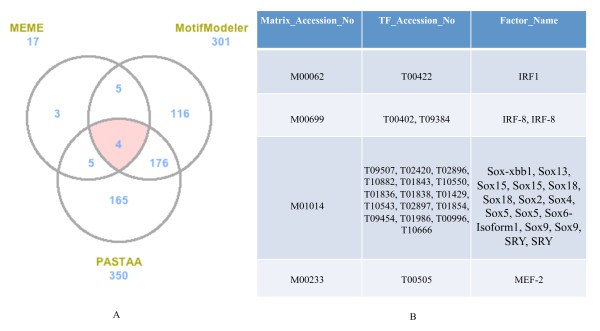
Comparison of predicted TFBS from MEME, MotifModeler, and PASTAA. A. Venn diagram showing numbers of predicted motifs from the three tools for the pro-inflammatory genes. The numbers next to the prediction tool names are the total numbers of top 70% predicted TFBS for each tool. B. List of common predicted motifs and the corresponding profile TFs. Profile TFs corresponding to these four common TFBS were obtained from TRANSFAC [15].
Comparison of TFBS prediction scores between test vs. random gene sets.
Matrix Accession Number |
TF name |
MEME |
MEME |
Motif Modeler |
Motif Modeler |
PASTAA |
PASTAA |
|---|---|---|---|---|---|---|---|
E-value* |
E-value** |
TCS score* |
TCS score** |
P-value* |
P-value** |
||
M01014 |
SOX |
1.60E+07 |
- |
1.49E-02 |
7.06E-03 |
2.99E-01 |
7.38E-01 |
M00699 |
IRF-8 |
6.90E-01 |
- |
1.50E-02 |
- |
3.50E-05 |
9.51E-01 |
M00233 |
MEF-2 |
1.60E+07 |
- |
1.54E-02 |
6.91E-03 |
5.75E-01 |
7.68E-01 |
M00062 |
IRF-1 |
2.60E-07 |
- |
1.55E-02 |
6.86E-03 |
7.40E-05 |
- |
Reciprocal comparison of TFBS prediction scores between random vs. test gene sets.
Matrix Accession Number |
TF name |
MEME |
MEME |
Motif Modeler |
Motif Modeler |
PASTAA |
PASTAA |
|---|---|---|---|---|---|---|---|
E-value* |
E-value** |
TCS score* |
TCS score** |
P-value* |
P-value** |
||
M00649 |
MAZ |
- |
4.10E+05 |
1.73E-02 |
8.28E-03 |
2.49E-02 |
6.92E-03 |
M00423 |
Foxj2 |
- |
1.80E+07 |
1.45E-02 |
7.25E-03 |
6.00E-01 |
5.05E-01 |
In the antibacterial gene set, the tools found 22, 7, and 178 motifs respectively with the intersection containing only one motif (Prrx2 as the profile TF). Due to the limited size of this latter data set, we did not compare prediction scores for this data with a random data set, as we did for the pro-inflammatory data. However, we performed target identification for Prrx2 using literature sources (next section).
Validation of TF-TFBS interactions controlling the pro-inflammatory genes
Biologically validated target genes of profile TFs predicted from the pro-inflammatory gene set.
Matrix Accession Number |
TF |
Target Genes |
|---|---|---|
M01014 |
Sox5 |
Smad5, Smad1, Smad7, Sox6, Sox5, Mir125b2, Mir34a, Mir224, Mir15a, Mir125b1, Lipe, Sry |
M01014 |
SRY |
Slc9a3r2, Wt1, Akr1b10, Zfp748, Hdac3, Smad3, Ar, Importin beta, Ep300, Kpnb1, Kpna, Znf208, Sry, Amh, Ptgds |
M00699 |
IRF-8 |
Spi1, Irf1, Trim21, Cops2, Irf2, Il12b, Il1b, Cybb, B2 m, Cbl, Irf4, Nfatc1, Ttraf6, Stat1, Etv6, beta2-mg, Cdkn2b, Il-12 p40, H-2Dd, H-2Kb, H-2Ld |
M01014 |
Sox13 |
Smad7, Fgf3 |
M01014 |
Sox4 |
Mir199a1, Mir27b, Mir199a2, Mir206, Mir29c, Mir107, Mir34a, Mir95, Mir17, Mir199b, Mirn292, Mirn101b, Cebpa, Sdcbp, Tcf4 |
M01014 |
Sox2 |
Pou5f1, Pou2f1, Pax6, Lbx1, Pdx1, Meis1, Asc, Golga6, Nkx2-3, Otp, Dlx5, Otx1, Dlx4, Isl1, Zfhx3, Fbxo15, Fgf4, Hrc, Nanog, Spp1, Zscan10 |
M01014 |
Sox9 |
Ep300, Nr5a1, Kpnb1, Crebbp, Smad3, Smad2, Amh, Mia, Med12, Maf, Importin beta, Calmodulin, Ppargc1a, Ncadherin, Col2a1 |
M01014 |
Sox15 |
Fhl3, Pou5f1 |
M00233 |
Mef-2 |
Smarca4, Hdac4, Hdac9, Hdac7, Hdac5, Nfat, Mapk14, Thra, Ep300, Ckm, Myog, Mef2 d, Jun, Slc2a4, Srf, Mck |
M00062 |
IRF-1 |
Agtr2, C2ta, Nos2, H-2kb, Ptgs2, Tlr3, H2-Dd, IL-12, IL-7R, Stat1, Ciita, Nfkb1, Rela, Stat5, Tap1, Vcam1, Psmb9, Ifnb1, Stat3, Irf8, Crebbp, Smarca4, Cybb |
M01014 |
Sox6 |
Cenpk, Sox5, Dazap2, Hdac1, Ctnnb1, Pdx1, Ctbp2, Mir29a, Mir221, Mir222, Mir29c, Mir126, Ccnd1, Fgf3, Hbb-y |
M01014 |
Sox18 |
Mef2c, Vcam1 |
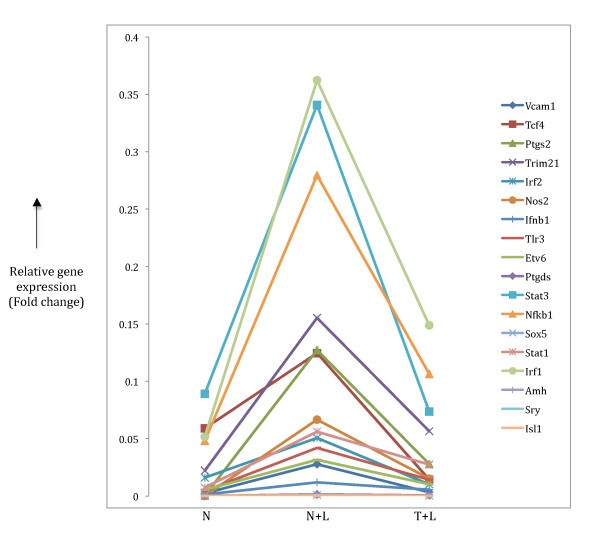
Expression patterns pro-inflammatory target gene. Target genes highlighted in Table 3 were checked to look for inflammatory gene expression pattern that is two-fold up regulation in N+L phenotype (compared to N) and further two-fold down regulation in T+L phenotype. The relative expression patterns of 18 target genes found in our microarray data are shown.
When we checked Prrx2, the only TF profile that was identified in our TFBS prediction from the antibacterial genes, it detected two genes (Ifi204, Pdgfra) as potential targets in the literature. Neither of these genes showed the antibacterial expression pattern in the Foster et al microarray data.
Discussion
As a basis for characterizing transcriptomic signatures in LPS tolerance, we differentiated pro-inflammatory and antibacterial gene expression in LPS-stimulated macrophages. The genes in the Foster et al [6] microarray data were classified into pro-inflammatory and antibacterial sets using biologically relevant filters (Figures 1 and 2). Authors of that study had performed similar categorization to identify "tolerizable" and "non-tolerizable" classes corresponding to our pro-inflammatory and antibacterial classes respectively. Basically, Foster et al and we hypothesize that during tolerance although pro-inflammatory genes are down regulated, antibacterial genes need to be continually expressed. The two classes in their study were shown to have differences in chromatin modifications indicating transient silencing of some pro-inflammatory genes while priming of antibacterial gene expression [6]. However, they did not attempt to identify any global TF-TFBS interactions that can attest to transcriptional signatures responsible for the distinction between the two classes and hence to LPS tolerance. We have identified and characterized such distinctive interactions employing pattern search algorithms combined with literature-based validation of the target genes in the microarray data.
Our filtering approach deducted only a small number (18) of antibacterial genes since the cut-off of even one-fold step-wise increase from the N to N+L to T+L stages along with the p-value threshold seems to be highly stringent and this may indicate a highly specific antibacterial phenotype for these genes. Due to the small size of this data set, subsequent analyses of motif search, and target identification and validation for this class of genes did not merit much attention. However, genes such as Lcn2 [18] and Tirap [19] in this list (Table S1) have shown antimicrobial activity. Interestingly Lcn2 suppressed LPS-induced inflammatory cytokines in macrophages [20] indicating an additional anti-inflammatory role for this gene. It is likely that a number of antibacterial genes in our microarray data maintain the same level of expression at both the N+L and T+L stages to provide persistent bacterial protection and hence it would be worthwhile to analyze a larger set of these genes by filtering with a less stringent condition.
Based on the concept of coordinate gene expression being controlled by same or similar TFs binding to their cognate binding motifs, we identified respectively four and one TFBS motifs in the upstream regulatory regions of the pro-inflammatory and antibacterial genes (Figure 4). The essence of the specificity of these TF-TFBS interactions was provided by the intersection of the predictions from three tools of differing algorithmic approaches. Additionally, when compared to a random gene background the prediction scores for the four pro-inflammatory motifs were significantly higher in the test data set (Tables 1 &2). Our predictions identified interferon regulatory factors 1 and 8 (IRF-1 and IRF-8) as pro-inflammatory TFs (Figure 4, Table 1) and these proteins have been clearly implicated in macrophage-associated innate immunity [21, 22, 23].
We validated the TF-TFBS predictions in the pro-inflammatory class of genes by manually identifying the corresponding target genes implicated to be under control of the predicted TFs. This analysis produced a significant number of target genes and a small proportion of them (18 out of 141) showed the pro-inflammatory specific pattern of gene expression (Table 3, Figure 5). A similar gene expression pattern of the 18 genes identified in our analysis with data from the Mages et al [10] microarray analysis (Table S4) confirms the genuine pro-inflammatory phenotype of these transcriptional targets in macrophage tolerance induction, and indicates that the four predicted TFs most likely control the transcriptional regulation of these genes to establish the phenotype. A number of genes in this list have been experimentally associated with LPS-mediated macrophage activation including some important TFs such as IRF1, IRF2, Stat1, Stat3 and Nfkb1 [8] that are essential members of presumptive transcriptional regulatory networks. Nfkb1 has been identified as a major player in the downstream signalling pathways of LPS-stimulated macrophages [24] with a crucial role in the transcriptional regulation of a number of target genes [9, 25]. More importantly, some of these target genes, such as Trim21, Ptgs2 and Nos2 (Figure 5) that show the sharpest drop in gene expression upon prolonged LPS treatment have been implicated in tolerance [26, 27, 28]. Nfkb1 acts as the controlling TF in LPS-induced expression of Ptgs2 and Nos2 [29] while acting downstream of Trim21 [26]. Here, since the microarray data represents a static view of gene expression, we cannot determine whether the Nfkb1 gene acts up or downstream of these genes as they all (Nfkb1, Trim21, Ptgs2 and Nos2) show the prototypical pro-inflammatory phenotype of down regulation (Figure 5) in induction of tolerance. A caveat to this classification may be the case of Nos2 that shows up in the pro-inflammatory category in our analysis. This gene has a direct role in killing intracellular pathogens [30, 31, 32] even though it shows down regulation in the tolerant state. A reason for the presence of such genes in our classification maybe due to the non-linearity of TF-TFBS interactions in that the dynamic modulation of transcriptional target gene expression does not always correlate with the corresponding TF binding to its site.
A limitation to this study is the lack of the tolerant (T) stage in the Foster et al [6] data set unlike the Mages et al [10] data. By employing the N+L stage instead of T (Figures 1, 2 and 5) we are likely to miss some genes (false negatives) that are tolerizable. However, the filtering approach that we employed is logically sound and did result in a significant number (228) of pro-inflammatory genes. Our motif prediction tools converged on 4 TF binding motifs that could co-ordinately regulate these genes (Figure 4). From a biological perspective, the 18 genes that are validated from the literature regards to being transcriptional targets of the four predicted profile TFs are most likely to be genuine candidates for establishing and/or maintaining the pro-inflammatory phenotype.
It is interesting to note that although the exhaustive work of Ramsey et al [8] looked at the dynamics of transcriptional programs in LPS-stimulated macrophages, they did not characterize the differential expression of the two categories of tolerant genes as belonging to the pro-inflammatory and antibacterial classes. We observed a number of common genes between our 228 pro-inflammatory set and their list of 1960 differentially expressed genes probably indicating a mixture of both classes of genes in their data. Litvak et al [9] have implicated one such gene, Cebpd, a TF, in a regulatory circuit discriminating between transient and persistent TLR4-stimulated signals. A search for the Cebpd binding motif in the regulatory regions of our two classes of genes (similar to their analysis) is likely to shed more light on the varying gene expression patterns in LPS tolerance induction.
Conclusions
By combining distinct gene expression array data with motif scanning and literature-based biological validation, we have identified and characterized transcriptomic signatures categorizing pro-inflammatory and antibacterial classes of genes in LPS tolerant macrophages. We identified 228 pro-inflammatory and 18 antibacterial genes likely to be transcriptionally regulated by four and one TFs respectively. Further, employing literature resources, we observed a number of target genes corresponding to the predicted profile TFs in the pro-inflammatory set and a subset of these targets clearly showed the pro-inflammatory gene expression pattern corresponding to LPS tolerance.
Methods
Differentially expressed genes
The Foster et al microarray dataset (GSE7348) was downloaded from the NCBI-GEO database [33]via FTP protocol. This data set was derived from murine (C57BL/6 strain) bone marrow macrophages left untreated (N), stimulated with LPS for 24 hours and then re-stimulated for 4 hours (T+L) or treated only with the second stimulation for 4 hours (N+L). RNAs from these 3 conditions were hybridized to the Affymetrix Mouse Genome 430 2.0 arrays (in duplicates) to get raw gene expression values for 45101 probe sets [6]. Differentially expressed genes were selected based on their expression level differences in various experimental conditions. For gene selection, in addition to fold change, we also performed Student's two-tailed t-test by assuming heteroscedasticity between the naïve and tolerant macrophages to prove significant differences in expression between the two conditions. For a pro-inflammatory gene, the ratio of average gene expression values in N+L to N and T+L to N+L should be > one and < one respectively, and have the t-test p-value of < 0.05. On the other hand, for an antibacterial gene, the ratio of average gene expression values in both N+L to N and T+L to N+L should be > one, and have the t-test p-value of <0.05. In this study, we utilized random mouse genes as background and they were selected using the RSAT tool [17]. The Mages et al [10] data was downloaded from GEO (GSE8621) and appropriate differential gene expression filters (based on fold change) were applied to compare with the pro-inflammatory transcriptional targets identified in our analysis of the Foster et al data.
Sequence retrieval
Regulator modules are generally located in the upstream and near the promoter regions of a gene. We considered this as our basis to retrieve the 1000 to +300 region, with respect to TSS, of all selected test and random genes. Gene sequences were retrieved from NCBI using a local tool [34] and repeats were masked using RepeatMasker [35].
Identification of TFBS
We used three TFBS prediction tools viz., MEME [12], MotifModeler [13], and PASTAA [14]. We performed this analysis to both our test and random sets of genes. Description and parameters used for these tools are as follows:
MEME
It works by searching for repeated, ungapped sequence patterns in the input DNA with statistical significance. The non-default parameters used for this analysis comprised of number of motifs (30), maximum width (15), and mode of motif distribution (zero or one per sequence, zoops). Additionally the revcomp parameter was used to search for TFBS in both strands. MEME identifies motifs without any concern to biological validation.
MotifModeler
Gene UIDs and corresponding expression values of a co-regulated set of genes were given as input and this software works by taking a set of random motifs of fixed size and mapping them onto putative regulatory regions of genes of interest. A linear model was established by considering the expression values and the efficacy of selected motifs. In this model each motif was evaluated based on its contribution to transcriptional regulation. This was iterated many times to calculate a cumulative transcription contribution score (TCS) that was used for motif selection. Least square method was used to dictate inhibitory and stimulatory effects of the predicted motifs. For the analysis of random gene data, we used random expression values, generated by a PERL script, within the range of the maximum and minimum expression values of test genes.
PASTAA
This software takes ENSEMBL gene ids as input and ranks them based on the annotated binding affinities of 549 vertebrate TRANSFAC TFs to their promoter regions. Simultaneously, it also ranks them based on their tissue specificities derived from expression data. These two lists are compared for high ranked common genes and an iterated hypergeometric test is performed to deduce the relationships between TFs and the list of genes and corresponding p-values calculated. Lower the p-value better the prediction.
Motif selection and their corresponding profile TFs
Top 70% of the predicted motifs were selected and compared in all tools. MEME predicts the consensus sequences of all possible hits in the promoter region of a given set of genes but does not provide any annotated information about the predictions. Hence, we took the position weight matrices (PWM) of the predicted consensus sequences and searched in TRANSFAC [15] for the motif accession numbers using the TOMTOM tool [36]. MotifModeler and PASTAA provide motif accession numbers for the predicted motifs. We used TRANSFAC to manually map these motifs to TFs.
Literature-based validation for target genes
We collected a list of genes (from TRANSFAC and IPA) that have been biologically identified as targets to the profile TFs that were discovered in motif prediction. We compared this list with the list of genes in the microarray that passed through a two-fold filter for the pro-inflammatory expression pattern. By normalizing the highest expression value in the N condition of the raw microarray data (45101 probe values) to 1, we calculated relative expression values for the filtered target genes corresponding to the N, N+L and T+L conditions (Figure 5).
Acknowledgements
We thank M. H. Kaplan for critical review of the manuscript. RKK was supported by a research fellowship from the IUPUI Graduate Office.
Declarations
Authors’ Affiliations
References
- Takeda K, Kaisho T, Akira S: Toll-like receptors. Annual Review of Immunology 2003,21(1):335.View ArticlePubMedGoogle Scholar
- Janeway CA, Medzhitov R: Innate Immune Recognition. Annual Review of Immunology 2002,20(1):197–216.View ArticlePubMedGoogle Scholar
- Karin M, Lawrence T, Nizet V: Innate Immunity Gone Awry: Linking Microbial Infections to Chronic Inflammation and Cancer. 2006,124(4):823–835.Google Scholar
- Liew FY, Xu D, Brint EK, O'Neill LAJ: Negative regulation of Toll-like receptor-mediated immune responses. Nat Rev Immunol 2005,5(6):446–458.View ArticlePubMedGoogle Scholar
- West M, Heagy W: Endotoxin tolerance: A review. Crit Care Med 2002,30(1 Supp):S64-S73.View ArticleGoogle Scholar
- Foster SL, Hargreaves DC, Medzhitov R: Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007,447(7147):972–978.PubMedGoogle Scholar
- Roach JC, Smith KD, Strobe KL, Nissen SM, Haudenschild CD, Zhou D, Vasicek TJ, Held GA, Stolovitzky GA, Hood LE, Aderem A: Transcription factor expression in lipopolysaccharide-activated peripheral-blood-derived mononuclear cells. Proceedings of the National Academy of Sciences 2007,104(41):16245–16250.View ArticleGoogle Scholar
- Ramsey SA, Klemm SL, Zak DE, Kennedy KA, Thorsson V, Li B, Gilchrist M, Gold ES, Johnson CD, Litvak V, Navarro G, Roach JC, Rosenberger CM, Rust AG, Yudkovsky N, Aderem A, Shmulevich I: Uncovering a Macrophage Transcriptional Program by Integrating Evidence from Motif Scanning and Expression Dynamics. PLoS Comput Biol 2008,4(3):e1000021.View ArticlePubMedGoogle Scholar
- Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich I, Aderem A: Function of C/EBP[delta] in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat Immunol 2009,10(4):437–443.View ArticlePubMedGoogle Scholar
- Mages J, Dietrich H, Lang R: A genome-wide analysis of LPS tolerance in macrophages. Immunobiology 2007,212(9–10):723–737.View ArticlePubMedGoogle Scholar
- MeV: MultiExperiment Viewer [http://www.tm4.org/mev.html]
- Bailey TL, Elkan C: Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 1994, 2:28–36.PubMedGoogle Scholar
- Liu Y, Taylor MW, Edenberg HJ: Model-based identification of cis-acting elements from microarray data. Genomics 2006,88(4):452–461.View ArticlePubMedGoogle Scholar
- Roider HG, Manke T, O'Keeffe S, Vingron M, Haas SA: PASTAA: identifying transcription factors associated with sets of co-regulated genes. Bioinformatics 2009, 25:435–442.View ArticlePubMedGoogle Scholar
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Land S, Lewicki-Potapov B, Michael H, Münch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E: TRANSFAC(R): transcriptional regulation, from patterns to profiles. Nucl Acids Res 2003,31(1):374–378.View ArticlePubMedGoogle Scholar
- Ingenuity Systems [http://www.ingenuity.com]
- Thomas-Chollier M, Sand O, Turatsinze J-V, Janky Rs, Defrance M, Vervisch E, Brohee S, van Helden J: RSAT: regulatory sequence analysis tools. Nucl Acids Res 2008,36(suppl_2):W119–127.View ArticlePubMedGoogle Scholar
- Saiga H, Nishimura J, Kuwata H, Okuyama M, Matsumoto S, Sato S, Matsumoto M, Akira S, Yoshikai Y, Honda K, Yamamoto M, Takeda K: Lipocalin 2-Dependent Inhibition of Mycobacterial Growth in Alveolar Epithelium. J Immunol 2008,181(12):8521–8527.PubMedGoogle Scholar
- Jeyaseelan S, Young SK, Yamamoto M, Arndt PG, Akira S, Kolls JK, Worthen GS: Toll/IL-1R Domain-Containing Adaptor Protein (TIRAP) Is a Critical Mediator of Antibacterial Defense in the Lung against Klebsiella pneumoniae but Not Pseudomonas aeruginosa. J Immunol 2006,177(1):538–547.PubMedGoogle Scholar
- Zhang J, Wu Y, Zhang Y, LeRoith D, Bernlohr DA, Chen X: The Role of Lipocalin 2 in the Regulation of Inflammation in Adipocytes and Macrophages. Mol Endocrinol 2008,22(6):1416–1426.View ArticlePubMedGoogle Scholar
- Dror N, Alter-Koltunoff M, Azriel A, Amariglio N, Jacob-Hirsch J, Zeligson S, Morgenstern A, Tamura T, Hauser H, Rechavi G, Ozato K, Levi B-Z: Identification of IRF-8 and IRF-1 target genes in activated macrophages. Mol Immunol 2007,44(4):338–346.View ArticlePubMedGoogle Scholar
- Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, Takayanagi H, Ohba Y, Taniguchi T, Honda K: Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proceedings of the National Academy of Sciences 2006,103(41):15136–15141.View ArticleGoogle Scholar
- Vila-del Sol V, Punzon C, Fresno M: IFN-{gamma}-Induced TNF-{alpha} Expression Is Regulated by Interferon Regulatory Factors 1 and 8 in Mouse Macrophages. J Immunol 2008,181(7):4461–4470.PubMedGoogle Scholar
- Dos Santos S, Delattre A.-I, De Longueville F, Bult H, Raes M: Gene Expression Profiling of LPS-Stimulated Murine Macrophages and Role of the NF-kB and PI3K/mTOR Signaling Pathways. Annals of the New York Academy of Sciences 2007, 1096(Signal Transduction Pathways Part D Inflammatory Signaling Pathways and Neuropathology)70–77.Google Scholar
- Kollet JI, Petro TM: IRF-1 and NF-[kappa]B p50/cRel bind to distinct regions of the proximal murine IL-12 p35 promoter during costimulation with IFN-[gamma] and LPS. Molecular Immunology 2006,43(6):623–633.View ArticlePubMedGoogle Scholar
- Yoshimi R, Chang T-H, Wang H, Atsumi T, Morse HC, Ozato K: Gene Disruption Study Reveals a Nonredundant Role for TRIM21/Ro52 in NF-{kappa}B-Dependent Cytokine Expression in Fibroblasts. J Immunol 2009,182(12):7527–7538.View ArticlePubMedGoogle Scholar
- Cao C, Matsumura K, Yamagata K, Watanabe Y: Involvement of cyclooxygenase-2 in LPS-induced fever and regulation of its mRNA by LPS in the rat brain. Am J Physiol Regul Integr Comp Physiol 1997,272(6):R1712–1725.Google Scholar
- Dias MB, Almeida MC, Carnio EC, Branco LGS: Role of nitric oxide in tolerance to lipopolysaccharide in mice. J Appl Physiol 2005,98(4):1322–1327.View ArticlePubMedGoogle Scholar
- Spitzer JA, Zheng M, Kolls JK, Vande Stouwe C, Spitzer JJ: Ethanol and LPS modulate NF-kappaB activation, inducible NO synthase and COX-2 gene expression in rat liver cells in vivo. Front Biosci 2002, 7:a99–108.View ArticlePubMedGoogle Scholar
- Utaisincharoen P, Anuntagool N, Arjcharoen S, Limposuwan K, Chaisuriya P, Sirisinha S: Induction of iNOS expression and antimicrobial activity by interferon-beta is distinct from IFN-gamma in Burkholderia pseudomalleiinfected mouse macrophages. Clinical & Experimental Immunology 2004,136(2):277–283.View ArticleGoogle Scholar
- Tarasenko O, Scott A, Soderberg L, Ponnappan U, Alusta P: Killing of Bacillus spores is mediated by nitric oxide and nitric oxide synthase during glycoconjugate-enhanced phagocytosis. Glycoconjugate Journal 2010,27(1):13–25.View ArticlePubMedGoogle Scholar
- Hazlett LD, McClellan S, Goshgarian C, Huang X, Thakur A, Barrett R: The Role of Nitric Oxide in Resistance to P. aeruginosa Ocular Infection. Ocular Immunology and Inflammation 2005,13(4):279–288.View ArticlePubMedGoogle Scholar
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R: NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucl Acids Res 2007,35(suppl_1):D760–765.View ArticlePubMedGoogle Scholar
- Grobe M: Get NCBI sequences for genes or specified regions. [http://discern.uits.iu.edu:8421/view-sequences.html]
- Smit AFA, Hubley R, Green P: RepeatMasker Open-3.0. 1996–2010. [http://repeatmasker.org]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS: Quantifying similarity between motifs. Genome Biol 2007,8(2):R24.View ArticlePubMedGoogle Scholar
- Beissbarth T, Speed TP: GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004,20(9):1464–1465.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.